|
Hina Patel
How difficult to watch your child suffer from “sickle cell disease” through frequent pain crises 18 Years?
NRI Couple, Bakerfield, CA, Hina Patel Foundation
Los Angeles, Aug 05, 2015/ NRIpress-Club/Ramesh/Gary Singh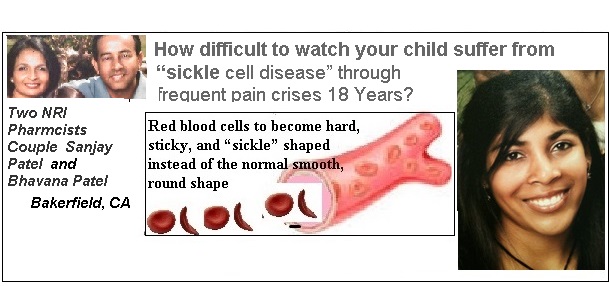
- Two NRI Pharmcists Couple Sanjay Patel and Bhavana Patel just married : Two weeks after their first child, Hina Patel was born in 1990, they received the startling news that her daughter, she had sickle cell disease….
- Every time she had a severe pain crises we wished there was a cure; the pain was unbearable for us to watch and more so for her to suffer
- Hina maintain her 4.1 GPA and accepted to the University of the Pacific Pharmacy Program, which sadly, she was never able to attend…..died on May 2010

——————————
Hina’s Story
by Hina’s Mother Bhavana Patel
It’s every parent’s dream to give birth to a healthy baby. When our daughter, Hina, was born on December 11, 1990, we were ecstatic to find that she was healthy. Things took a turn for the worst when we received a letter from the hospital informing us that Hina had been diagnosed with a blood disorder known as “hemoglobinopathy.” Her blood was tested by electrophoresis, and she was diagnosed with Sickle Beta Thalassemia. We were immediately convinced that the diagnosis was wrong because she showed no immediate signs of the symptoms. However, when Hina experienced hand-foot syndrome on her first birthday, our deepest fears were confirmed. To watch Hina cry relentlessly from the pain, and know that all we could do at the time was give her Motrin, made us feel so helpless. Soon, she began receiving blood transfusions every two months to keep her hemoglobin high. We were transferred to a hematologist at (Children’s Hospital Los Angeles) CHLA because of the possibility of an iron overload from the frequent transfusions. At CHLA, the doctors recommended against giving any more transfusions unless Hina’s hemoglobin dropped below 7g/dl or if she had trouble breathing.
Hina would experience severe pain in her hips, legs, and arms every three to four months and be hospitalized or provided with home health care. She was not able to walk, move or eat during these crises, and would run fevers as high as 105 degrees. We lived in apprehension because her crises would occur unexpectedly. Our lives revolved around taking care of Hina, making sure she took her medications, drank at least 10 glasses of water per day, didn’t overexert herself, stayed warm, but not too hot, and kept her away from sick children.
In 2001, what started out as a cold raged into a severe pain crises and acute chest syndrome. Hina was transported via helicopter to Children’s Hospital Central California for treatment. As my husband and I drove to the hospital in Madera and arrived at 2 AM, we were terrified we might lose her. Her oxygen capacity was at 38% and she was placed on the ventilator (mechanical breathing machine). It took her two months to completely recover before she could return back to school. Three months later, she was readmitted to the hospital with a severe pain crisis. Her hematologist started her on Hydroxyurea, which helped most of her pain crises for about four years. However, even on Hydroxyurea, she still experienced pain in her abdomen and when she entered high school, she had her gallbladder removed due to gallstones. Despite the surgery, the abdominal pain continued, so she went to see a GI specialist. Soon after, she was treated for H-pylori and placed on Prevacid. The treatment didn’t help and Hina’s pain episodes continued throughout high school. The stress of high school and extracurricular activities made her pain episodes more frequent and just plain unbearable.
As a parent it was difficult to watch our child suffer as much as she did. This led my husband to search for a bone marrow transplant match in hopes of one day curing her disease. We sought the opinions of two expert transplant hospitals: Lucille Packard Hospital of Stanford and Children’s Hospital of Los Angeles. None of our family members, including her sister were a match. To our great pleasure, a closely matched unrelated donor was found and Hina received her bone marrow transplant on May 31, 2008 at CHLA. She responded well in the beginning, but as months grew into a year, she was bogged down with Adenovirus infection, C-difficile colitis, and graft versus host disease (GVHD) of the skin, colon and liver. After 24 long months of isolation, on May 5, 2010, Hina passed away due to liver failure.
Throughout her life, no one would have guessed Hina suffered from Sickle Cell Anemia just by her outward appearance. Although she was a child with hemoglobin of 7 g/dl, Hina was always active and creative. Hina didn’t like watching TV or playing videogames as other children did. She loved to go bike riding with her dad and play sports with friends. Her wild imagination was unceasingly portrayed in her crafts, plays, and games created for the family and friends. Baking, traveling, decorating for holidays, and learning about new things were what she enjoyed doing the most. Most impressively, throughout her pain and suffering , education was a top priority for her; she studied hard and tried to get ahead with her homework so that she would not fall behind during her multiple cycles of pain crises. Even during her months in the isolation, she continued her learning, excelled in her AP courses, and was accepted into the University of the Pacific. She never lost sight of her future goal to one day become a pharmacist.
Hina lived in an isolation room throughout the transplant process for months at a time and wasn’t allowed to go outside. She never once complained about not being able to soak up the sunlight, feel the breeze, or cold ocean water.
Hina was incredibly selfless. A few months before receiving her transplant, she worked to create a Sickle Cell Awareness fair in her community to provide support to those suffering from the disease. She hoped to let others know that they are not alone and that together, they could pull through. Despite her pain, she was able to put others first and the continuation of the Sickle Cell Awareness Fair in her memory is the least we can do. More importantly, I know that she would want it to succeed to provide others with the help and support that they need.
Hina’s smile never left her face throughout this entire process. She was brave, strong, and determined. She is an inspiration to all those who knew her, and made us all realize how much meaning the little things in life hold. There are no words to express her loss; the only comfort we have now is knowing that she is protected in a better place finally at peace from the pain.
Hina’s Bone Marrow Journey
As Hina’s mother, I know that it is difficult to watch your child suffer through frequent pain crises. It is a helpless feeling, and naturally, as a mother, you want to do all that you can for them. The reason I want to share Hina’s story is to give other parents going through a similar situation a clear view of what receiving a bone marrow transplant may entail. There are numerous complications to be aware of and the procedure is extremely risky. Still, there is always hope that the transplant may be a success.
Hina was diagnosed with Sickle Cell Beta Thalassemia 3 weeks after birth. Every time she had a severe pain crises we wished there was a cure; the pain was unbearable for us to watch and more so for her to suffer. The multiple complications of Sickle Cell Disease clouded the minds of my husband and I and led us to look for different alternatives. My husband came across the option of a bone marrow transplant. Since our other child was not an HLA match, he looked for an unrelated donor. An HLA-typed match was found (9 out of 10): a 42 year-old male donor. We sought the opinions from two expert transplant hospitals: Children’s Hospital of Los Angeles and Lucille Packard Children’s of Stanford and were told there could be a success rate of approximately 80-85%. Complications such as GVHD and infections were also discussed and we were also informed that the procedure could possibly be life threatening. We spoke to two patients with Sickle Cell Disease who had undergone bone marrow transplants and survived with very few complications, which made us hopeful. Prior to the transplant, Hina had a complete check up of all her organs and was deemed healthy enough to undergo the procedure. Hina underwent the bone marrow transplant on May 31, 2008 at CHLA. She had 10 days of conditioning with chemotherapy to ablate her existing marrow cells. Hina tolerated the chemotherapy regimen without any problems and the new donor marrow was infused. Two weeks later, her white blood cells, including the neutrophils, red blood cells, and platelets counts were on the rise and she was completely engrafted with 100% donor cells. She had no more Sickle Cell Disease, the news we had hoped for. After 45 days of dealing with headaches and hypertension, she developed rashes on her arms and upper back. This was her first sign of skin GVHD and she was placed on treatment with Prednisone. After 60 days of staying in an isolation room, Hina was sent home. We were very happy, but soon more complications developed and we were making visits to CHLA one or two times every week. Over the next two years after the transplant, Hina was in and out of the hospital for a cumulative duration of 9 months in isolation. We lived in constant fear, not knowing what problem we would have to face in the next minute. We traveled to the Seattle Children’s Hospital on December 27, 2010 in hopes that they would be able to cure our daughter. They made many adjustments to her GVHD medications, which further complicated the problem. Since Seattle was too far to travel for medical attention needed on daily basis, we sought medical care at Mattel Children’s Hospital at UCLA. She was treated by the Pediatric Blood and Marrow Transplant team for 4 months, but the complications of her liver GVHD became so severe that nothing more could be done and she passed away on May 5, 2010 at UCLA.
• Chemotherapy Medications used for conditioning- Busulfan, Cyclophsophamide, Fludarabine, and Alemtuzumab—Complications—Adenovirus in blood and stool, GVHD of skin rash & loss of hair/ diarrhea EVERY day for months, which eventually resulted in bloody diarrhea/, yellow eyes & skin, 7-10 diarrhea with blood daily, Hypertension (high blood pressure), Hyperglycemia (high blood sugar), Hypothyroid, Pancreatitis, swelling due to water retention, collection of blood outside of a blood vesse, leg cramps, headaches, anorexia, Weight gain due to prednisone, Osteoporosis, Hypercholeserolemia (high cholesterol), electrolyte imbalance, abdominal pain, gas, and countless others.
• Invasive Procedures-Hickman Catheter, Picc Line, ndoscopy/Colonoscopy 4 times, Bone Marrow Biopsy 4 times, Skin Biopsy, Liver Biopsy, Intubation for Angiogram, Cauterization on the cecum area of colon.
• Lab tests 3-4 times per week, going to home health care 3 times per week for IV meds, daily infusing of IV meds & TPN at home, going 3 to 4 times per week for platelet and blood transfusions, changing picc line dressing weekly, taking 60 to 90 pills per day, many trips to the emergency room.
• Total Platelet Transfusions over 2 years: 129 times/Total Blood Transfusions over 2 years: 44 times—
Our brave daughter went through an indescribable amount of suffering, but never once did she complain about any of it. She kept a positive attitude, smiled continuously, and studied in the isolation room to maintain her 4.1 GPA and high AP exam scores. She graduated high school and was determined to become a pharmacist. She applied and was accepted to the University of the Pacific Pharmacy Program, which sadly, she was never able to attend.
If I, as a mother, had a second chance, I would not have decided to put my daughter through the transplant process. While there might be a chance of a cure, there is also the possibility that the child will be worse off than they were to begin with. Bone Marrow Transplant is a hope for a cure but not one that should be considered lightly. No one, including physicians, has a crystal ball (the ability to see the future) to predict the outcome for any given individual. I implore every family considering a Bone Marrow Transplant to research and explore every option and seek out multiple medical opinions before making such a life changing decision. For more information please feel free to contact me.
There are 250 million people in the world that carry the sickle cell gene,” said Sanjay Patel, who explained that children are often diagnosed at birth. “It’s a very cruel disease, you just never know what’s going to happen.”
Sickle cell anemia is a lifelong genetic disorder that originated as a prevention mechanism from Malaria, according to Sanjay Patel. As a reaction, the gene becomes defective and causes the normal blood cells to die too quickly. As the damaged sickle cells travel through the body they then become stuck, cutting off the oxygen from other parts of the body and causing them to die, he said.
These are the realities that those with sickle cell anemia face every day. In fact, stress, changes in the weather and a busy lifestyle can all trigger the adverse reactions of the sickle cells, according to Sanjay Patel.
“I hope she’s smiling up in heaven and saying, ‘OK, at least my parents are doing a little bit what I was thinking,'” Sanjay said.
————————————————————–
Indian-American couple to raise funds for sickle cell research
Washington, Sep 4, 2015:
A charity organization run by an Indian-origin couple will start a fundraising campaign on Saturday to benefit sickle cell disease research in the US, a media report said on Thursday.
“There are 250 million people in the world that carry the sickle cell gene,” the Bakersfield Californian quoted Sanjay Patel as saying on the radio and live video streaming programme “First Look with Scott Cox” on Thursday.
His wife Bhavana Patel also featured on the show.
Patels are the board members of Hina Patel Foundation, a charity that helps individuals suffering with sickle cell disease and their families by raising awareness, providing support group, and raising funds for research.
“It is a very cruel disease, you just never know what is going to happen,” Sanjay Patel said.
The fundraising event has been planned at Bakersfield’s Riverwalk Park and the proceeds will go to benefit Sickle Cell research at organisations like University of California, Los Angeles (UCLA) Medical Centre.
Bhavana and Sanjay founded their charity in 2010 after watching sickle cell anaemia ravaging their daughter Hina’s fragile body.
Sickle cell anaemia is a lifelong genetic disorder that originated as a prevention mechanism from Malaria, according to Sanjay Patel.
As a reaction, the gene becomes defective and causes the normal blood cells to die too quickly.
As the damaged sickle cells travel through the body they then become stuck, cutting off the oxygen from other parts of the body and causing them to die, he said…..IANS

|